These analyses suggest that this individual’s depression may be a manifestation of cytokine-induced interference with the conversion of tryptophan to melatonin and serotonin in conjunction with increased glutamate receptor activation.
Background
Recent studies have focused on the role of immune dysfunction in depression. and analogies to “cytokine-induced sickness behavior”.
Sickness Behavior – Behaviors that develop during the course of an infection such as lethargy, depression, sleep disturbances, and social withdrawal. These are generally evolutionarily helpful to enhances recovery by conserving energy.
How it Works
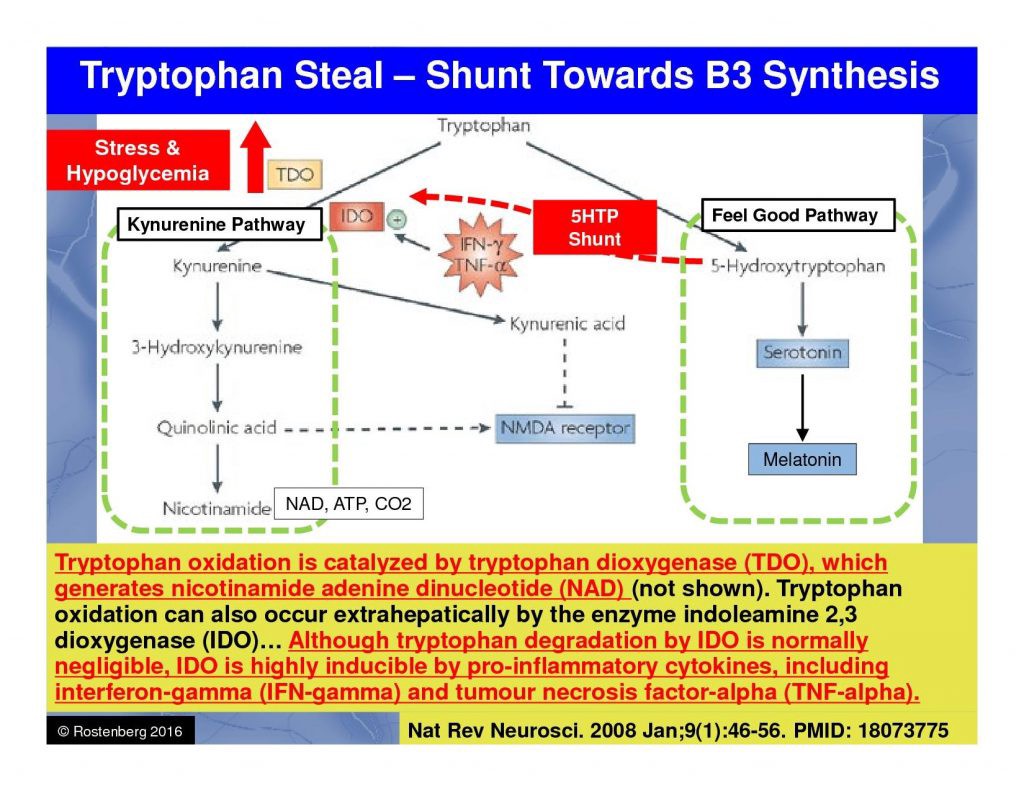
- Inflammation – An immune activation including increased production of proinflammatory cytokines has repeatedly been described in major depression. Proinflammatory cytokines such as interleukin-2, interferon-γ, or tumor necrosis factor-α activate the tryptophan- and serotonin-degrading enzyme indoleamine 2,3-dioxygenase (IDO).
- Less Serotonin/Melatonin Ingredients – Depressive states during inflammatory somatic disorders are also associated with increased proinflammatory cytokines and increased consumption of tryptophan via activation of IDO.
- Lower Serotonin – Enhanced consumption of serotonin and its precursor tryptophan through IDO activation could explain the reduced availability of serotonergic neurotransmission in MD.
- Anxiety-Related Receptor Activation – Besides the well-known deficiency in serotonergic neurotransmission as a correlate of major depression (MD), recent evidence points to a pivotal role in increased glutamate receptor activation as well. Increased activation of IDO and its subsequent enzyme kynurenine monooxygenase by proinflammatory cytokines, moreover, leads to enhanced production of quinolinic acid, a strong agonist of the glutamatergic N-methyl-D-aspartate (NMDA) receptor.
Case Presentation
Introduction
This 39-year-old male presented for the treatment of treatment-resistant unipolar major depression without psychotic symptoms. Additional comorbid symptoms include:
- Skin Conditions Such Including Psoriasis
- Pain – Inflammatory joint pain, knee swelling, and back pain
- Mood – anhedonia, anxiety, unipolar depression
- Digestive Issues
Complaint Details
- Loss of interest in daily activities
- Lack of energy
- Inability to focus (attention-deficit disorder)
- Irregular sleeping patterns
- Loss of appetite
- Low or decreased self-worth and self-esteem
- Suicidal thoughts
- Mild autistic symptoms
Onset & History:
- Age 8 – 39: Depression, anxiety
- Age 15 – 35: acne on face and back
- Age 15 – 35: Severe social anxiety
- Age 29 – 39: plaque psoriasis
- Age 15 – 35: back pain
- Age 39 – 39: guttate psoriasis
- Age 29 – 39: episodic arthritis
Quick Inventory of Depressive Symptomatology (QIDS)
Patient scored 21 points on QIDS inventory suggesting a state of severe depression.
Previous Treatments
Selective Serotonin Reuptake Inhibitors (SSRIs)
- 40 mg/day Citalopram (Celexa) for 6 months – No response, no remission
- 20 mg/day Escitalopram (Lexapro) for 3 months – No response, no remission
- 60 mg/day Fluoxetine (Prozac) for 6 months – No response, no remission
- 50 mg/day Paroxetine (Paxil) for 3 months – No response, no remission
- 100 mg/day Sertraline (Zoloft) for 9 months – No response, no remission
Serotonin and Norepinephrine Reuptake Inhibitors (SNRIs)
- 60 mg/day Cymbalta (duloxetine) – No response, no remission
- 150 mg/day Effexor (venlafaxine) – No response, no remission
Tricyclic Antidepressants (TCAs)
- 100 mg/day Anafranil – No response, no remission
Monoamine Oxidase Inhibitors (MAOIs)
- 10 mg/day Eldepryl (Selegiline Hcl) – Moderate response, no remission
Atypical Antidepressants
- 30 mg/day Remeron (mirtazapine): Moderate response, no remission
- 300mg/day Wellbutrin (bupropion): Moderate response, no remission, increased energy
Related Factors
Inflammation
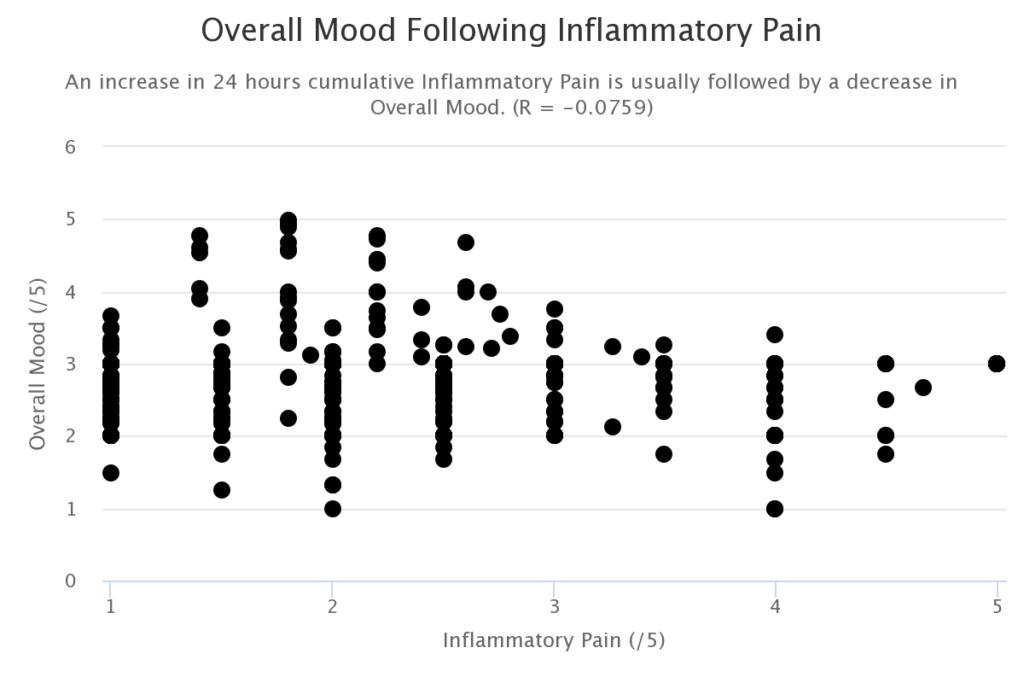
Analysis of regular self-reported pain and mood measurements by the patient reveals a strong correlation between inflammatory pain and severity of depressive symptoms. This is consistent with the hypothesis that elevated inflammatory cytokine levels may be initiating the shunt of tryptophan away from intracranial serotonin production as depicted in Figure 1. Tryptophan shunt away from serotonin/melatonin production to neurotoxic quinolinic acid.
Sleep
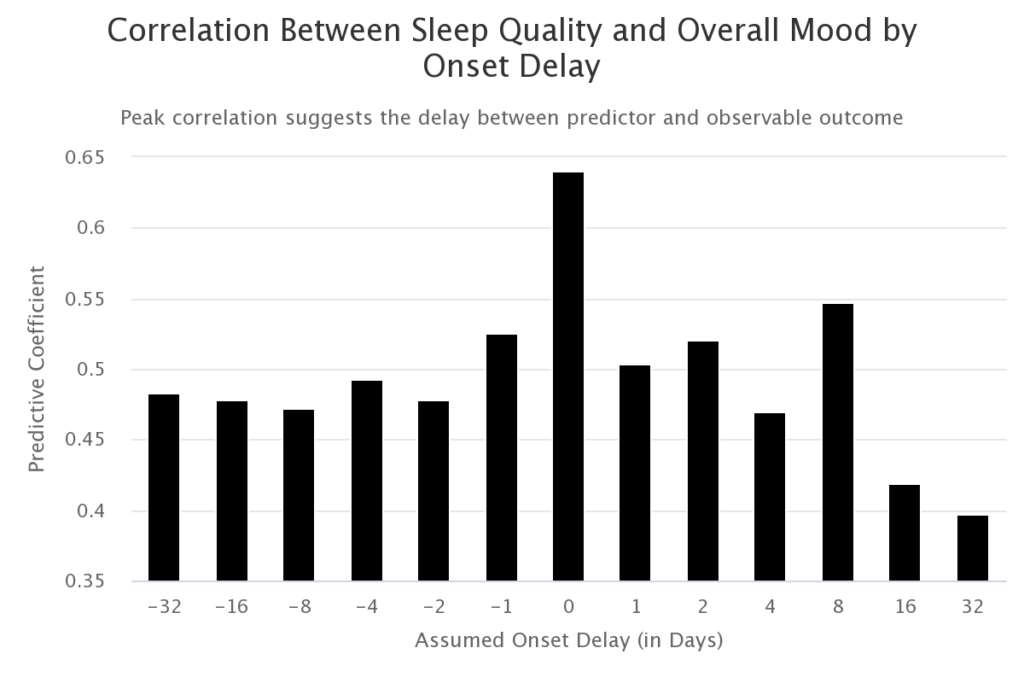
Sleep quality was calculated based on the amount of movement during sleep as measured by a Fitbit accelerometer. Melatonin is the main hormone involved in the control of the sleep-wake cycle. Cytokine-induced depletion of serotonin and melatonin (Figure 1) is further supported by a strong correlation between the patient’s sleep quality and mood the preceding day.
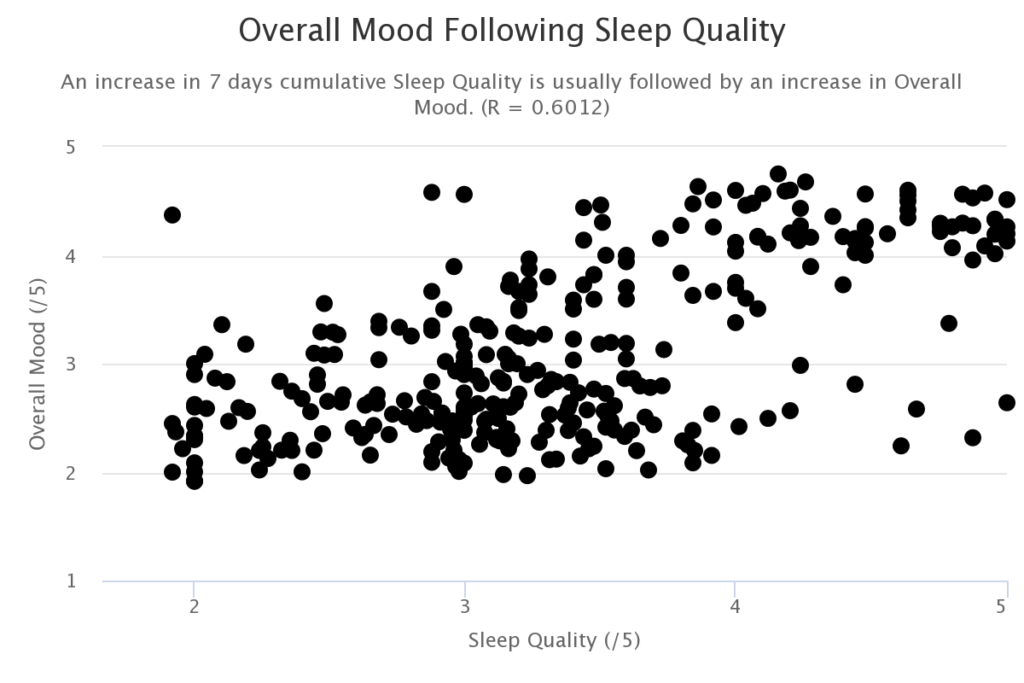
The fact that the correlation between mood and sleep quality is highest without pairing the sleep quality measurements with mood outcome data lagged by a day suggests that the relationship is correlational rather than causal. This is illustrated in the following chart where the correlation at a 0-day lag is much higher than the correlation at a 1-day lag.
Diet
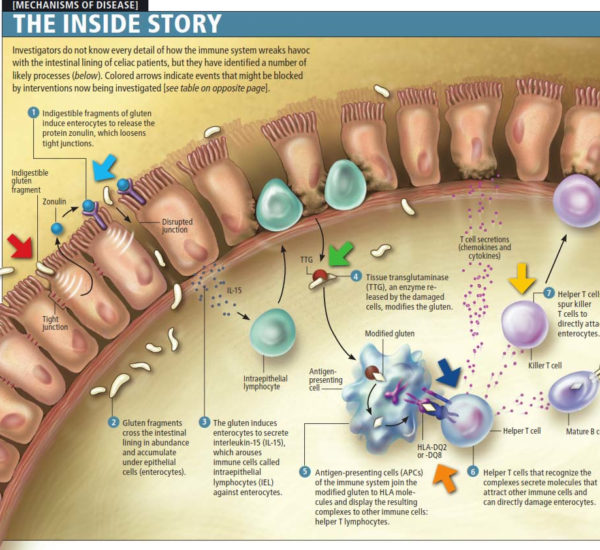
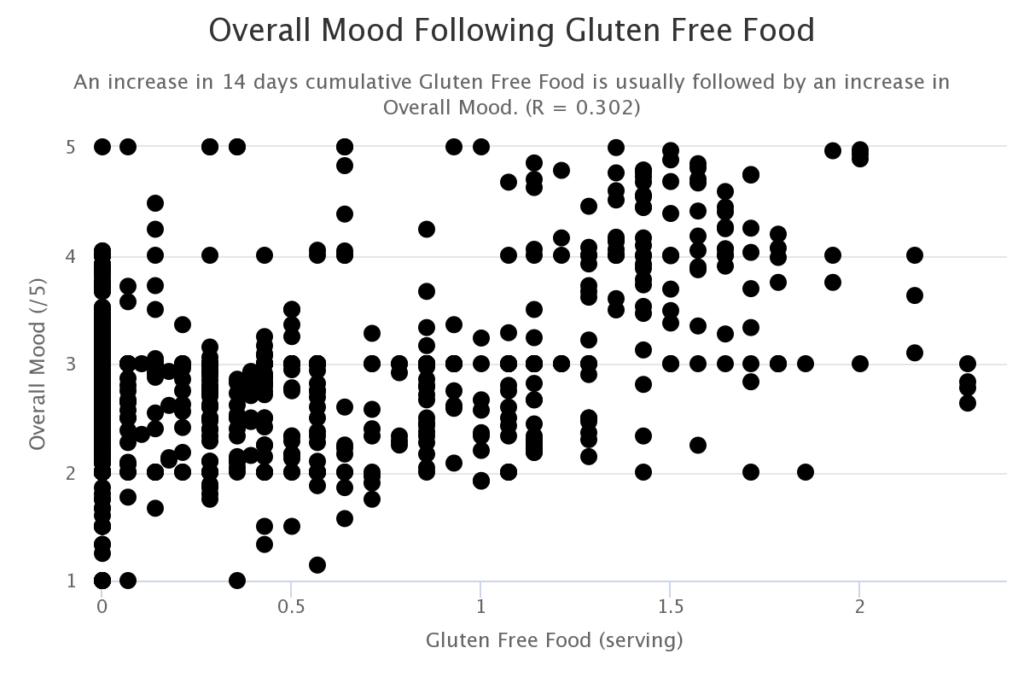
Acne, social anxiety, and inflammatory back pain were completely alleviated following the removal of gluten from the subject’s diet at age 35. Several attempts at the reintroduction of gluten result in the recurrence of acne, social anxiety, and inflammatory pain within 2 days and gradually subside over the following 2 weeks after the last gluten exposure.
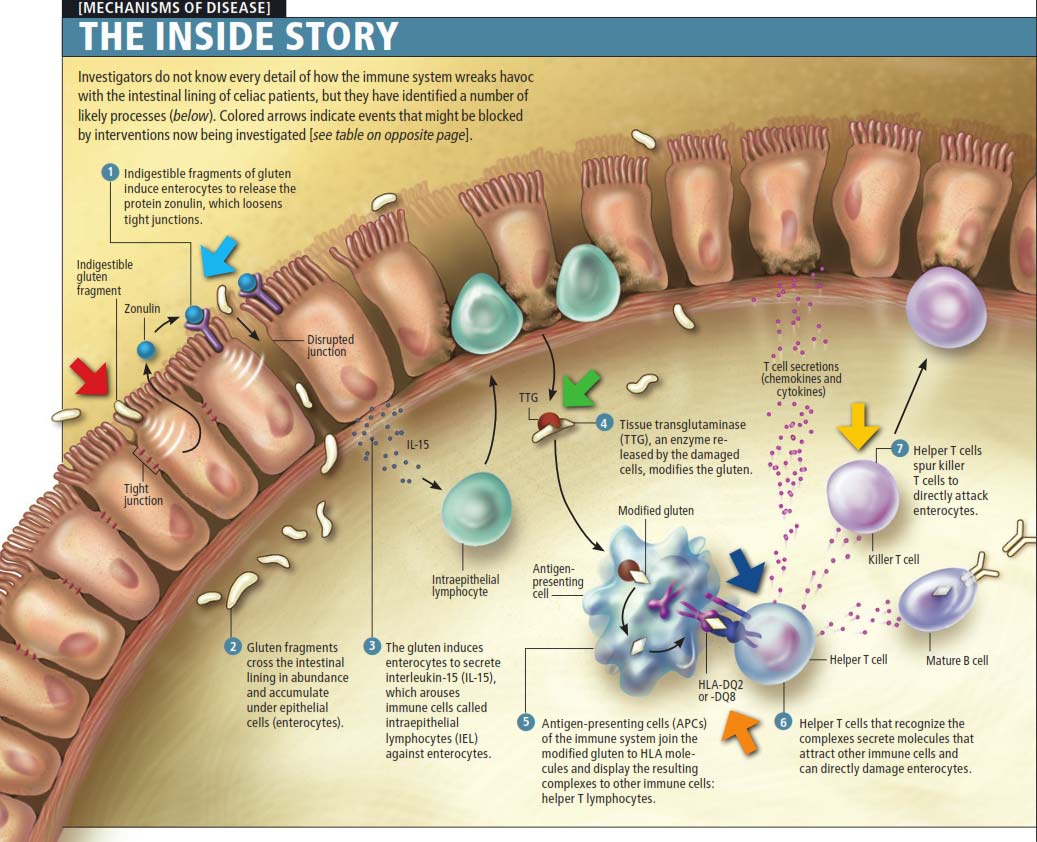
It has been shown that gluten can trigger the release of the protein zonulin. Zonulin then loosens the tight junctions in the intestinal wall. This allows toxins to enter the bloodstream triggering an immune response. This leads to the elevation of the cytokines which interfere with the conversion of tryptophan to serotonin and melatonin.
Psoriasis is a systemic autoimmune inflammatory disease that shares some immunological aspects with other inflammatory-based diseases, such as Crohn’s disease.1 Bacterial DNA (bactDNA) fragments have been shown to induce a systemic immunological response in Crohn’s disease and other settings. The translocation of gut bacteria to the bloodstream is also associated with the increase in inflammatory mediators that can lead to inflammatory and neuro-inflammatory disease.
| Inflammatory Mediator (mean pg/mL) | BactDNA-Positive Psoriasis (n = 16) | BactDNA-Negative Psoriasis (n = 38) | P |
| Interferon γ | 130.8 | 29.7 | <.001 |
| Tumor necrosis factor | 95.2 | 21.7 | <.001 |
| Interleukin 1β | 68.3 | 13.6 | <.001 |
| Interleukin 6 | 176.8 | 36.9 | <.001 |
| Interleukin 12 | 125.9 | 38.8 | <.001 |
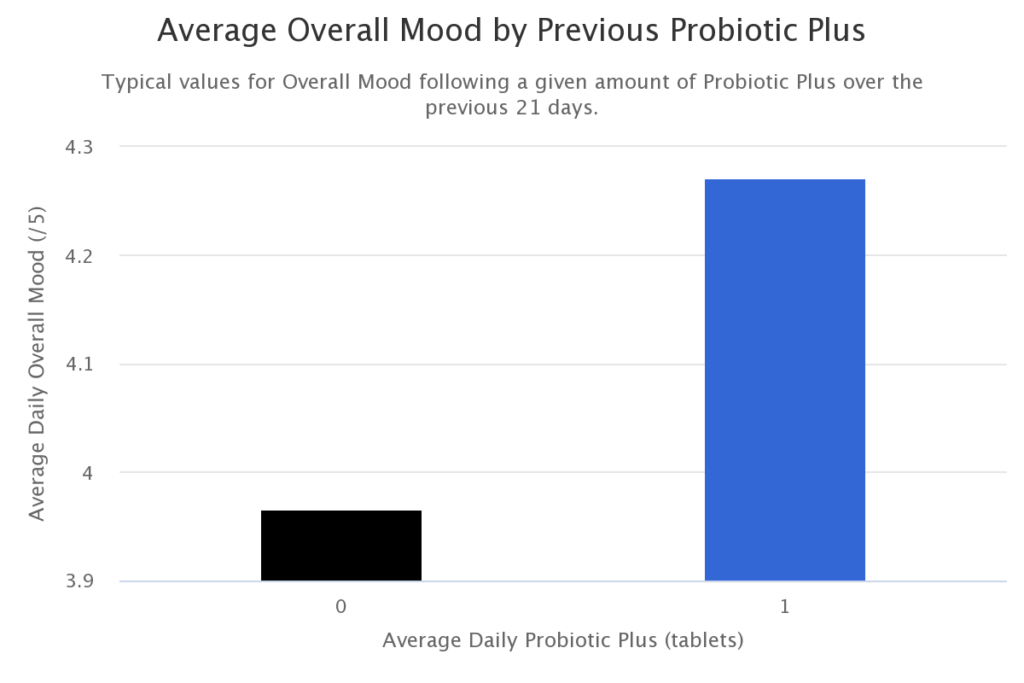
Probiotics
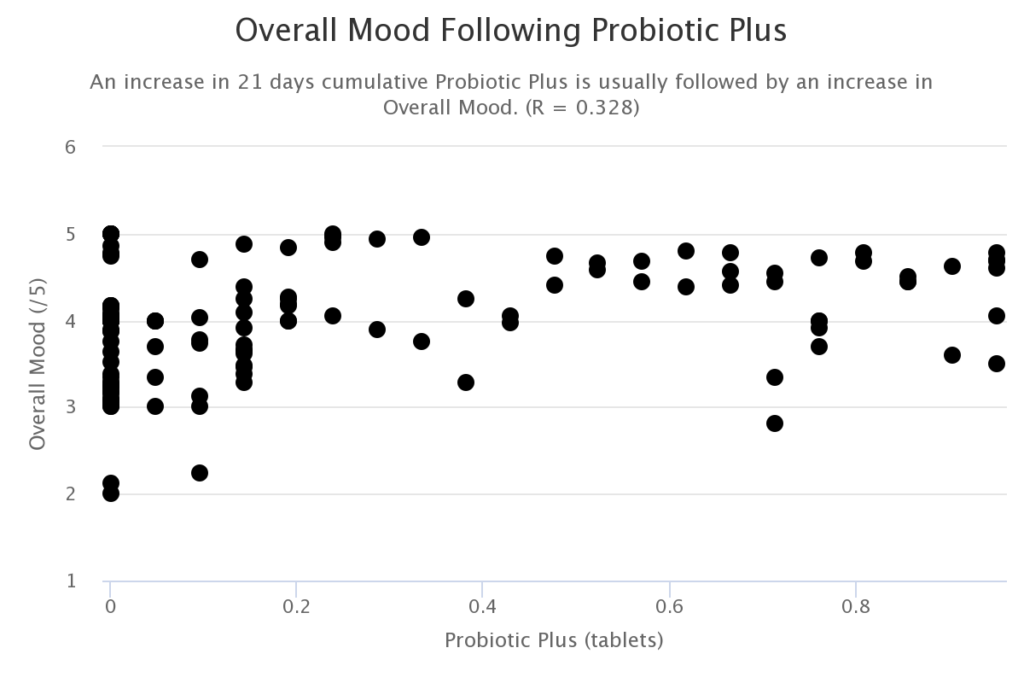
Intake of the probiotic Bacillus coagulans (Probiotics Plus) is predictive of elevated overall mood. It has been shown in mouse models that Bacillus coagulans combined with a prebiotic can reduce intestinal permeability and associated inflammatory reactions.
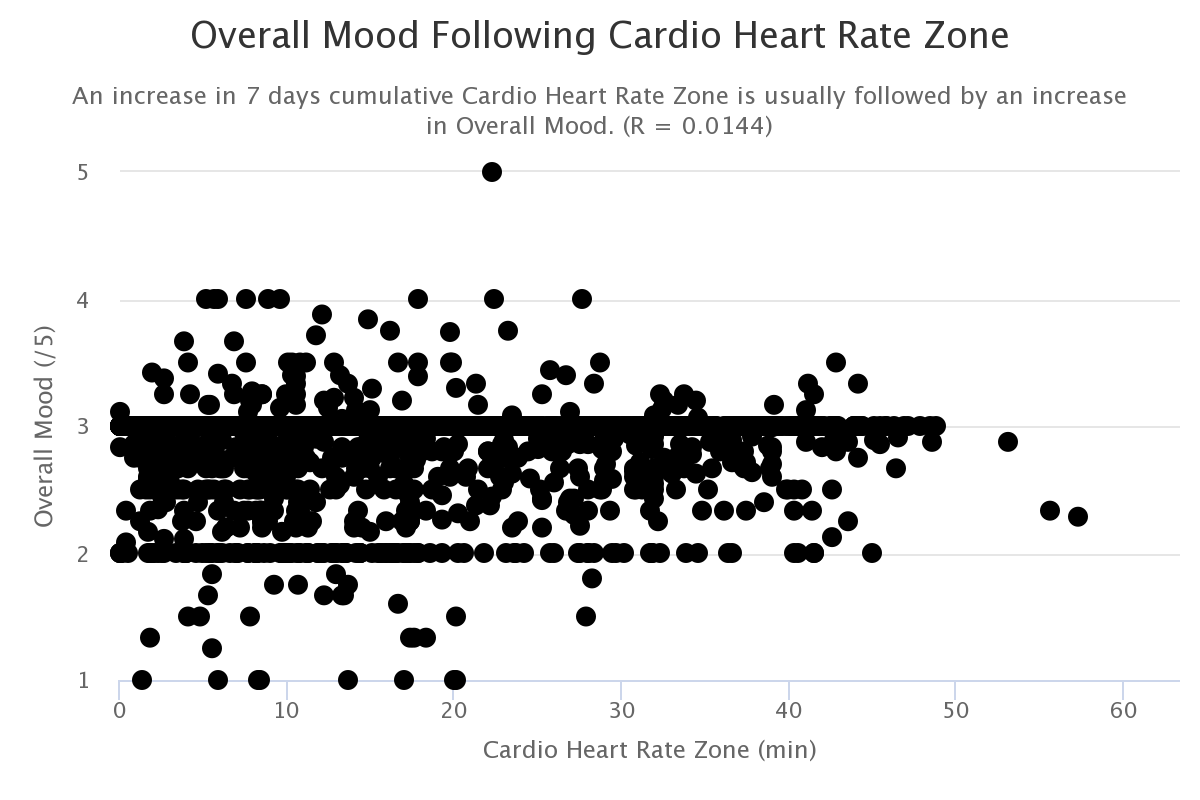
Exercise
Physical activity appears to provide lesser medium-term benefits.
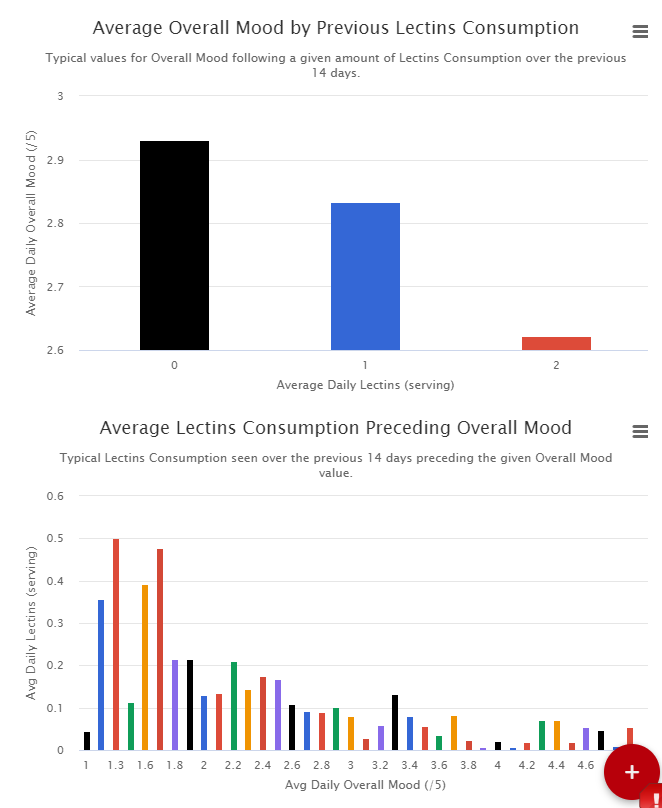
Lectins
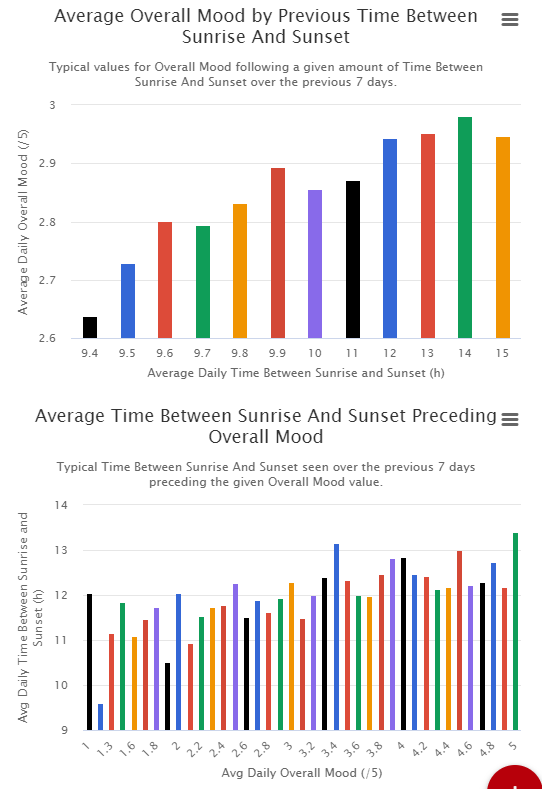
Sunlight
Likely Prognosis or Disease Progression
N-methyl-D-aspartate receptors (NMDAR) overactivation is linked to neurodegeneration. The current prevailing theory suggests that synaptic and extrasynaptic NMDAR (syn- and ex-NMDAR) impose counteracting effects on cell fate, and neuronal cell death is mainly mediated by the activation of ex-NMDAR.
N-methyl-D-aspartate receptors (NMDARs) mediate excitatory neural transmission. In addition to their physiological functions, mounting evidence has suggested their involvement in excitotoxicity. Cell death triggered by seizure or ischemic stroke is attributed to NMDAR overactivation.1, 2 Abnormal NMDAR activity is also associated with neurodegenerative disorders, such as Alzheimer’s, Huntington’s, and Parkinson’s disease.3 Thus, it is concluded that, while appropriate NMDAR activation is essential for neuronal survival and physiological functions, excessive activation contributes to pathological changes including cell death.4
The most plausible mechanism of action is related to inflammatory cytokine-induced NMDA ecotoxicity. Inflammation has been widely observed to significantly increase with aging. This suggests that the severity of the patient’s depression and suicidal ideation may only increase without intervention such as the NMDA-receptor antagonist Spravato to address the potential mechanism of action.
Autism
Mild. Diagnosis based on psychometric evaluation at https://psychcentral.com/quizzes/autism-test/
Based upon your responses to this autism screening measure, it appears that you are likely suffering from an autism spectrum disorder or Asperger’s disorder. People who score similarly often qualify for a diagnosis of autism or Asperger’s.
People with an autism spectrum disorder often suffer from severe and sustained impairment in social interaction and the development of restricted, repetitive patterns of behavior, interests, and activities. The disturbance must cause clinically significant impairment in social, occupational, or other important areas of functioning.
Genetic Markers
If you have your DNA sequenced, here’s a good free site for analyzing it: https://codegen.eu
https://www.geneticlifehacks.com/23andme-raw-data/
MTHFR Mutations
The MTHFR gene codes for an enzyme called methylenetetrahydrofolate reductase. This enzyme is critical for many different functions, including folate metabolism [18, 29], breaking down homocysteine [18], and making epigenetic changes to DNA through methylation [93]. Several common mutations in MTHFR have been associated with an increased risk of depression and other health problems [18, 93, 94, 95]. Carriers of certain SNP variants in the MTHFR gene have reduced activity of methylenetetrahydrofolate reductase [96, 97, 18, 98, 99]. This can result in folate deficiencies, elevated homocysteine levels, and reduced levels of important neurotransmitters — each of which can worsen mood [18, 100, 101, 102].
C677T MTHFR Mutation
Status: POSITIVE
This patient possesses the C677T homozygous A/A genotype mutation for the rs1801133 SNP marker of the MTHFR gene. This mutation (the A allele) is associated with reduced enzyme activity, elevated total homocysteine levels and altered distribution of folate [1]. People with an “AA” allele for this mutation present a 70% decrease of the normal enzyme activity [5].
Low levels of active folate or vitamin B12 can therefore lead to elevated blood homocysteine levels (a condition known as hyperhomocysteinemia) [110] . High levels of homocysteine are linked to a wide variety of health problems, including depressive moods [18, 107, 111]. Elevated homocysteine may contribute to mood problems by interfering with the production of important neurotransmitters, such as dopamine, norepinephrine, and serotonin [110, 18, 95].
A1298C MTHFR Mutation
Status: NEGATIVE
This patient possesses the normal T/T genotype for the rs1801131 SNP marker of the MTHFR gene. Thus, he does not possess the A1298C mutation. This mutation also impacts the MTFHR activity and the homocysteine levels but to a lesser extent than C677T [1]. 7% to 12% in North America, Australia and Europe have the 23andme MTHFR A1298C mutation.
See https://you.23andme.com/tools/data/?query=MTHFR&filter_by_platforms=false
https://selfhacked.com/blog/need-know-mthfr-genespolymorphisms-c677t-rs1801133/
CNR1 rs1049353 Genotype: C/C
Variation in the cannabinoid receptor type 1 (CNR1) gene may also contribute to risk for PTSD, with the minor (A) allele of rs1049353 associated with an increased likelihood of PTSD.6 Additionally, rs1049353 has been found to interact with childhood physical abuse (CPA), one type of trauma that might impact the endocannabinoid system, to predict anhedonia.7
The T allele reduced the development of depression when exposed to stressful life events, whereas those with the C allele experienced less pleasure in life (anhedonia) (R).
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4783167/
[rs324420]
The SNP rs324420 is part of the “bliss” gene FAAH, which is involved in breaking down anandamide (the main natural cannabinoid in the brain). Your genotype (‘AC’; heterozygous) is associated with increased activity of this gene, suggesting that you have an elevated risk of low mood [11]. Having higher FAAH gene activity means that cannabinoids — your natural “bliss molecules” — are broken down more quickly, reducing the overall activity of your brain’s endocannabinoid system. Because this system plays a key role in counteracting the stress response and creating positive moods, carriers of risk alleles in these genes are more likely to experience mood problems [74]. Therefore, you may be able to counteract your risk by inhibiting the FAAH gene, as well as by increasing the activity of your cannabinoid system in general.
Cannabidiol (also known as CBD) is a great option for doing both of these. CBD reduces activity of the FAAH gene, which means that your brain’s natural cannabinoids will stick around longer, and will be more effective in enhancing your mood and feelings of bliss [75]. CBD also stimulates the production of cannabinoid (CB1) receptors, which will provide additional benefits to your mood [76]. Another effective way to counteract elevated FAAH gene activity is to increase your brain’s levels of important cannabinoids, such as anandamide. Therefore, it would be a good idea to engage in regular exercise, which has been shown to improve mood by stimulating the release of anandamide throughout the brain [77].
Management and Outcome
Recommended Testing
- Folate and homocysteine
- methylmalonic acid (MMA) and holoTranscobalamin(holoTC)
- Therefore, it would be a good idea to check your levels of these important compounds, as well as your levels of vitamins B6 and B12.
- https://www.secondopinionphysician.com/shop/walsh-protocol-lab-test-histamine-methylation/
- In addition to iron and cobalamin (B12) deficiencies, other micronutrient deficiencies include deficiencies of water-soluble vitamins (e.g., thiamin and nicotinamide) and decreased absorption of fat-soluble vitamins (vitamins A, D, E, and K).
Treatment:
Inflammation Reduction
Suggestions based on SelfDecoded DNA analysis:
- Take a resveratrol supplement or eat foods high in it, especially dark-colored berries (See your SNPs: General Inflammation, Dermatitis, and Arthritis)
- Consider drinking chamomile tea (See your SNPs: General Inflammation)
- Try supplementing with curcumin as it helps reduce inflammation (See your SNPs: Crohn’s Disease)
- Ensure you follow a rigorous daily dental hygiene regimen (See your SNPs: Periodontitis)
Depression Reduction
Top three health recommendations made in SelfDecoded DNA analysis:
- Make sure you’re getting plenty of regular exercises.
- Look into getting more cold exposure, such as by taking cold showers or ice baths.
- Take cannabidiol (CBD oil)
Sources
https://journals.physiology.org/doi/abs/10.1152/ajpgi.00245.2020?journalCode=ajpgi
https://www.sciencedirect.com/science/article/abs/pii/S0269749119336991
https://www.frontiersin.org/articles/10.3389/fpsyt.2018.00205/full
https://www.mdpi.com/1424-8247/11/3/63/htm
https://jneuroinflammation.biomedcentral.com/articles/10.1186/1742-2094-8-94
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5997870/
https://bmcpsychiatry.biomedcentral.com/articles/10.1186/s12888-014-0372-y
https://pubmed.ncbi.nlm.nih.gov/30213652/
https://pubmed.ncbi.nlm.nih.gov/23438698/
https://www.nature.com/articles/4002006
https://jamanetwork.com/journals/jamadermatology/fullarticle/2174889